Компьютерный анализ процессов структурообразования нуклеиновых кислот
|
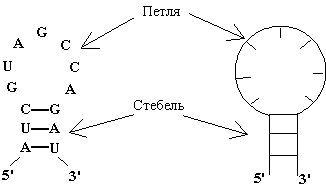
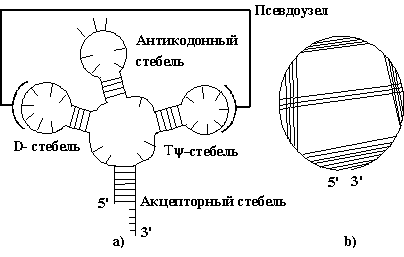
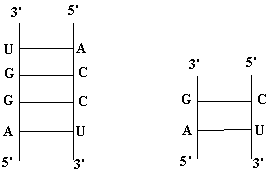
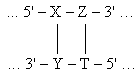
|
XY – ZT |
|
XY – ZT |
|
XY – ZT |
|
|
AU-AU |
–0.9 |
CG-AU |
–2.1 |
GC-CG |
–3.4 |
|
UA-AU |
–1.3 |
GC-AU |
–2.4 |
GC-GC |
–3.3 |
|
CG-UA |
–2.0 |
GC-UA |
–2.2 |
|
|
|
AU-UA |
–1.1 |
CG-GC |
–2.4 |
|
|
Таблица 6.1. Свободная энергия элементарных
ячеек (в килокалориях на моль).
Асимметрия стэкинг-взаимодейчствия. Из таблицы видно, что энергии ячееек
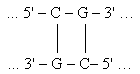 и
и 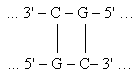
(т.е. CG-GC и GC-CG) существенно различны
(-2.4 и –3.4), хотя ячейки отличаются друг от друга только направлениями нитей.
Это дает нам пример того, что стэкинг-взаимодействие существенно зависит от
ориентации нитей двойной спирали.
Свободная энергия петли. Энергия петли
более точно называется энергией инициации петли. Она зависит от типа петли и от
ее длины. Длиной петли называется число неспаренных нуклеотидов в ее
однонитевых участках. Энергии коротких петель (до 10 нуклеотидов) даются в
специальных таблицах, полученных экспериментально. Энергия более длинных петель
определяется по формуле
![]() (6.1)
(6.1)
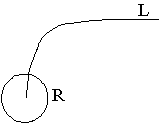
Рис. 6.3. Молекулярная нить
длины ![]() и
сфера Утсон-Криковского взаимодействия эффективного радиуса
и
сфера Утсон-Криковского взаимодействия эффективного радиуса ![]() .
.
Пусть
петля имеет физическую длину L=mN (где m - коэффициент пропорциональности).
Зафиксируем один конец петли. Тогда второй конец может находиться в точках шара
объемом ![]() . Обозначим за
. Обозначим за ![]() эффективный
радиус сферы Уотсон-Криковского взаимодействия. Тогда
эффективный
радиус сферы Уотсон-Криковского взаимодействия. Тогда ![]() – ее объем.
Вероятность попадания свободного конца петли в эту сферу можно оценить как
– ее объем.
Вероятность попадания свободного конца петли в эту сферу можно оценить как ![]() . Из формулы Гиббса (4.1) имеем
. Из формулы Гиббса (4.1) имеем 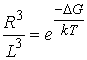 . Прологарифмировав это равенство, получим искомый закон изменения
свободной энергии петли в зависимости от ее длины.
. Прологарифмировав это равенство, получим искомый закон изменения
свободной энергии петли в зависимости от ее длины.
Энергия
инициации петли. Свободная энергия петель часто называется энергией их инициации.
Следует отметить, что, как правило, она положительна, т.е. сами по себе петли
неустойчивы и существуют только за счет отрицательной энергии стеблей, на
которые они опираются.
7.
Модель
процесса структурообразования.
Наш подход состоит в
математическом моделировании двух основных черт транскрипционного процесса:
последовательного роста молекулярной цепи РНК в ходе транскрипции и
постепенного последовательного формирования ее вторичной и третичной структуры.
В основу модели образования вторичной структуры
РНК положено взаимодействие двух основных процессов, влияющих на
структурообразование. Первый, элонгация, это последовательный рост молекулярной
цепи в ходе транскрипции. Второй, структуризация, есть последовательное
возникновение и формирование вторичной структуры РНК на том участке ее
молекулярной цепи, который уже образовался к данному времени. Структурные
перестройки при этом обеспечивают локальную минимизацию свободной энергии
сформировавшегося участка молекулы. Оба этих процесса рассматриваются как
дискретные. Молекулярная цепь удлиняется на целое число нуклеотидов, вторичная
структура изменяется путем возникновения или разрыва целого числа вторичных
связей. Отметим, что основным параметром в такой модели является относительная
скорость элонгации T, определяемая как отношение скорости
роста молекулярной цепи к скорости структурообразования (параметр структуризации).
В рамках нашей модели параметр T можно рассматривать как количество новых
нуклеотидов, на которое удлинится молекулярная цепь РНК за то время, пока на
старом ее участке будет происходить формирование и стабилизация вторичной
структуры.
Длина стебля = 1 Длина стебля
= 3
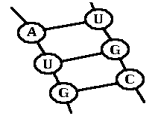
Рис. 7.1. Стебли являются
элементарными блоками, из которых строится вторичная структура РНК.
а)
б) в)
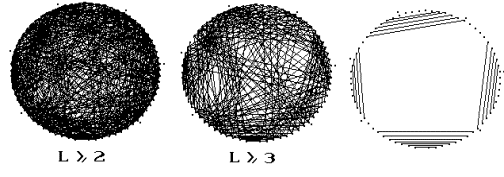
Рис.7.2. Транспортные РНК. Конкурирующие
стебли длины L для аспарагиновой тРНК человека
(а-б) и ее естественная вторичная структура - клеверный лист (в).
|
Длина |
Число стеблей |
Способность
описывать структуры |
|
|
893.3 |
100% |
|
|
270.5 |
98.7% |
|
|
109.9 |
95.1% |
Рис.7.3. Точность описания структуры падает
при увеличении минимально допустимой длины стеблей L (приведены средние
значения для транспопртных РНК).

Рис. 7.4. Разнообразие структурных элементов
резко растет при увеличении точности описания вторичной структуры, т.е. при
уменьшении минимально допустимой длины стеблей L.
Взаимодействие между указанными процессами
моделируется в простейшей форме. Считается, что фазы роста молекулярной цепи и
структуризации разделены во времени и циклически повторяются одна за другой. Скорости
обоих процессов полагаются постоянными и не зависящими от нуклеотидного состава
молекулярной цепи РНК, от сложности образующейся структуры и других
обстоятельств. При таких допущениях модель роста вторичной структуры РНК в ходе
транскрипции выглядит следующим образом.
Процесс описывается как пошаговый. На каждом шаге
сначала происходит структуризация, при которой вторичная струкутура РНК формируется
в пределах того участка молекулярной цепи, который уже образовался к данному
шагу процесса. После образования и стабилизации вторичной структуры происходит
удлинение молекулярной цепи на постоянное число нуклеотидов, равное параметру
структуризации процесса ![]() . После этого
происходит переход к следующему шагу процесса. Процесс заканчивается после
того, как молекулярная цепь вырастет полностью и структура полностью
сформируется.
. После этого
происходит переход к следующему шагу процесса. Процесс заканчивается после
того, как молекулярная цепь вырастет полностью и структура полностью
сформируется.
Известно, что некоторые молекулы РНК могут образовываться
в составе более длинного транскрипта, включающего их. Чтобы отразить этот факт
в модели, используется еще один параметр ![]() - начальная длина молекулярной цепи. Это
число нуклеотидов молекулярной цепи, уже образовавшихся к началу первого шага
процесса.
- начальная длина молекулярной цепи. Это
число нуклеотидов молекулярной цепи, уже образовавшихся к началу первого шага
процесса.
Таким образом, на шаге процесса с номером ![]() молекулярная цепь содержит
молекулярная цепь содержит ![]() нуклеотидов. На последнем шаге процесса
величина
нуклеотидов. На последнем шаге процесса
величина ![]() ограничивается полной длиной молекулярной
цепи, обозначаемой
ограничивается полной длиной молекулярной
цепи, обозначаемой ![]() . Поскольку оба
параметра процесса,
. Поскольку оба
параметра процесса, ![]() и
и ![]() , целочисленные,
то, исходя из физического смысла, мы имеем
, целочисленные,
то, исходя из физического смысла, мы имеем ![]() ,
, ![]() . Если
. Если ![]() , то полное
развитие процесса происходит уже на первом шаге и результат совпадает с
результатом для случая
, то полное
развитие процесса происходит уже на первом шаге и результат совпадает с
результатом для случая ![]() . Поэтому
поведение процесса образования вторичной структуры РНК моделируеися в области
. Поэтому
поведение процесса образования вторичной структуры РНК моделируеися в области ![]() ,
, ![]() на дискретной решетке
на дискретной решетке ![]() ;
; ![]()
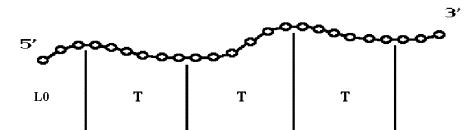
Рис. 7.5. Процесс
сворачивания РНК во вторичную структуру: Последовательный, пошаговый, с локальной
минимизацией свободной энергии на каждом шаге. Начинаясь с ![]() нуклеотидов,
длина цепи РНК увеличивается каждый раз на T, до тех пор, пока
не будет достигнут конец цепи. Основные параметры процесса: a) набор термодинамических
параметров – модель свободной энергии; b)
нуклеотидов,
длина цепи РНК увеличивается каждый раз на T, до тех пор, пока
не будет достигнут конец цепи. Основные параметры процесса: a) набор термодинамических
параметров – модель свободной энергии; b) ![]() – длина начального участка цепи РНК на первом
шаге
– длина начального участка цепи РНК на первом
шаге ![]() ; c)
; c) ![]() (период
структуризации) – длина участка, добавляемого к растущей цепи РНК на каждом
шаге.
(период
структуризации) – длина участка, добавляемого к растущей цепи РНК на каждом
шаге.
Опишем теперь более подробно, как моделируется
структуризация, т.е. процесс формирования вторичной структуры РНК на том ее
участке, который уже образовался. Это тоже пошаговый процесс. К началу
структуризации молкулярная цепь может уже обладать вторичной структурой,
возникшей на предыдущих шагах основного процесса. В нашей модели структура
наращивается путем добавления к ней элементарных структурных элементов -
стеблей. Стебель – это двуспиральный участок вторичной структуры, состоящий из
комплементарно спаренных оснований без пропусков. При добавлении к структуре
нового стебля, в ней возникают новые вторичные связи и могут разрываться
старые. Модель структуризации включает в себя правила формирования множества
допустимых стеблей и правила определения межструктурного перехода. В наиболее
широком варианте множество допустимых стеблей включат в себя все стебли, оба
спаренных участка которых находятся в пределах той части молекулярной цепи,
которая имеется к данному моменту времени. В этом случае возможно не только
наращивание вторичной структуры (т.е. возникновение новых вторичных связей), но
и ее перестройка, т.е. разрушение старых связей, препятствующих возникновению
новых. В более узком варианте множество допустимых стеблей содержит только
стебли, наращивающие структуру без необходимости разрыва старых связей. В этом
случае вторичная структура формируется без перестроек. Заметим, что множество
допустимых стеблей можно рассматривать как вариант множества допустимых
межструктурных переходов. В настоящей работе мы будем рассматривать только
модель процесса с разрешением любых структурных перестроек.
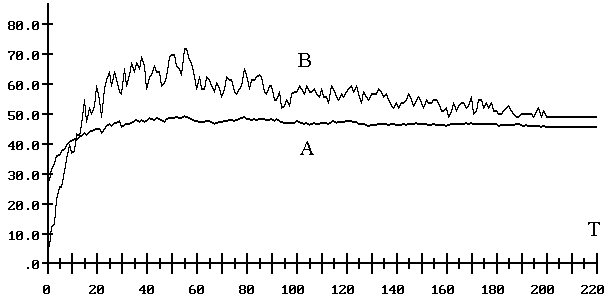
Рис. 7.6. Расчет вторичной
структуры 106 молекул РНК-ферментов. Качество предсказания
в зависимости от скорости приращения молекулярной цепи. Плавный график (A) – это средний
процент правильно предсказанных связей вторичной структуры. Прерывистый график
(B) – это средняя доля молекул, у которых вычисленная
вторичная структура не менее чем на 50% совпадает с натуральной. В дапазоне
скорости роста молекулярной цепи от 20 до 60 нуклеотидов за раз набдюдается
заметный рост качества предсказания вторичной структуры.
Как уже упоминалось, структуризация моделируется
как пошаговый процесс. На каждом ее шаге формируется множество допустимых
стеблей. Затем выбирается такой стебель, добавление которого к структуре даст
наибольшее понижение ее свободной энергии. Этот стебель добавляется к
структуре, после чего повторяется следующий шаг структуризации. Процесс
кончается после того, как структура стабилизируется. Это означает, что
множество тех допустимых стеблей, добавление которых способно понизить
свободную энергию структуры, пусто. Иначе говоря, процесс кончается, когда мы
попадаем в точку локального минимума свободной энергии вторичной структуры на
множестве допустимых межструктурных переходов.
После окончания процесса структуризации молекулярная
цепь удлинняется и цикл шагов основного процесса повторяется.![]()
8.
Простейшая
модель биополимера.
Будем
называть биополимер однородным, если он состоит из одинаковых элементов, и
нейтральным, если эти элементы не взаимодействуют друг с другом (кроме
полимерной связи соседних элементов). В этом разделе мы покажем, что простейшей
моделью однородного нейтрального биополимера является винтовая линия (спираль).
При этом мы рассмотрим самый простейший идеальный случай, который никогда не
достигается в реальности. Мы будем считать, что молекула биополимера «висит» в
абсолютно пустом пространстве при отсутствии внешних сил. Допустим, что
биополимер состоит из последовательности совершенно одинаковых элементов. Будем
моделировать элемент твёрдым телом. Поскольку все элементы одинаковые, то
каждый следующий элемент прикрепляется к предыдущему совершенно одинаковым
образом. Формально это описывается
следующим образом. К элементу привязывается сопутствующая система координат.
Положение и ориентация сопутствующей системы полностью определяет положение
элемента в пространстве. Обозначим ![]() начало сопутствующей
системы элемента
начало сопутствующей
системы элемента ![]() в абсолютной системе
координат. Рассмотрим ломаную линию
в абсолютной системе
координат. Рассмотрим ломаную линию ![]() ,
, ![]() ,…,
,…, ![]() ,…, вершины которой расположены в началах сопутствующих
систем элементов биополимера. Утверждение, которое мы хотим доказать, состоит в
следующем.
,…, вершины которой расположены в началах сопутствующих
систем элементов биополимера. Утверждение, которое мы хотим доказать, состоит в
следующем.
Утверждение. Возможны только три
случая
a) Точки ![]() ,
, ![]() ,…,
,…, ![]() ,… лежат на некоторой прямой, разбивая её на одинаковые
отрезки.
,… лежат на некоторой прямой, разбивая её на одинаковые
отрезки.
b) Точки ![]() ,
, ![]() ,…,
,…, ![]() ,… лежат на некоторой окружности, разбивая её на одинаковые
дуги.
,… лежат на некоторой окружности, разбивая её на одинаковые
дуги.
c) Точки ![]() ,
, ![]() ,…,
,…, ![]() ,… лежат на некоторой винтовой линии, разбивая её на
одинаковые сегменты.
,… лежат на некоторой винтовой линии, разбивая её на
одинаковые сегменты.
Доказательство. Пусть к элементу A прикреплён следующий элемент B. Тогда сопутствующая система B получается из A сдвигом и ортогональным преобразованием. По теореме Шаля ортогональное
преобразование представляет собой поворот вокруг некоторой оси (проходящей
через начало системы B). Поскольку
элементы одинаковы, то вектора сдвигов и поворотов в их сопутствующих системах
одинаковы. В абсолютном пространстве
вектор сдвига уже может меняться при переходе от одного элемента к другому. А
вот вектор поворота постоянен и в абсолютном пространстве. В самом деле, при
вращении системы координат вокруг какой-либо оси проекции её направляющего
вектора на оси координат не меняются. Поэтому координаты вектора поворота
последующего элемента в системе текущего элемента совпадают с его координатами
в системе предыдущего элемента. По той же причине проекция вектора сдвига на
вектор поворота в абсолютном пространстве постоянна. Вектор поворота в
абсолютной системе координат обозначим ![]() , а угол поворота
, а угол поворота ![]() .
.
Если поворот нулевой, то мы
получаем случай a). Каждая следующая точка ![]() получается из
предыдущей сдвигом на постоянный вектор.
получается из
предыдущей сдвигом на постоянный вектор.
Пусть поворот ненулевой, но
вектор поворота ортогонален вектору сдвига. В этом случае все вектора сдвига и,
следовательно, точки ![]() будут лежать в
плоскости, проходящей через
будут лежать в
плоскости, проходящей через ![]() и ортогональной
вектору поворота. Поскольку длины векторов сдвига одинаковы и эти вектора
поворачиваются на один и тот же угол, то мы получаем случай b).
и ортогональной
вектору поворота. Поскольку длины векторов сдвига одинаковы и эти вектора
поворачиваются на один и тот же угол, то мы получаем случай b).
Рассмотрим теперь общий
случай. Фактически нам достаточно доказать, что в абсолютном пространстве
найдутся ось, V параллельная вектору поворота ![]() , и вектор W, параллельный оси V, такие, что для любого i отрезок
, и вектор W, параллельный оси V, такие, что для любого i отрезок ![]() получается из отрезка
получается из отрезка
![]() поворотом вокруг оси V на постоянный угол
поворотом вокруг оси V на постоянный угол ![]() и сдвигом на вектор W. Заметим, что поскольку вектор W параллелен оси V, то порядок сдвигов и поворотов
несущественен.
и сдвигом на вектор W. Заметим, что поскольку вектор W параллелен оси V, то порядок сдвигов и поворотов
несущественен.
Возьмём
какую-либо плоскость ![]() , ортогональную вектору поворота
, ортогональную вектору поворота ![]() . Спроектируем нашу ломаную на эту плоскость. В качестве
вектора W возьмём проекцию
вектора сдвига на вектор поворота
. Спроектируем нашу ломаную на эту плоскость. В качестве
вектора W возьмём проекцию
вектора сдвига на вектор поворота ![]() в абсолютном
пространстве. Поскольку она постоянна, то проекция ломаной
в абсолютном
пространстве. Поскольку она постоянна, то проекция ломаной ![]() ,
, ![]() ,…,
,…, ![]() ,… на плоскость
,… на плоскость ![]() состоит из одинаковых
отрезков, повёрнутых на постоянный угол
состоит из одинаковых
отрезков, повёрнутых на постоянный угол ![]() друг относительно
друга. Значит, точки проекции ломаной лежат на некоторой окружности и ось V проходит через её центр и параллельна
вектору поворота
друг относительно
друга. Значит, точки проекции ломаной лежат на некоторой окружности и ось V проходит через её центр и параллельна
вектору поворота ![]() .
.
Доказательство
завершено.
Полученный
в данном разделе результат качественно можно трактовать следующим образом.
Биополимерам, состоящим из однородных элементов, должно быть свойственно
образовывать спиральные структуры. Как мы увидим далее, это действительно имеет
место как для белков, так и для нуклеиновых кислот.

Рис. 9.1. Тонкий упругий
стержень в свободном состоянии.
9.
Пространственная
структура.
Математическая модель двуспиральной молекулы ДНК на
основе тонкого упругого прямолинейного однородного симметричного стержня стала
уже классическим средством для изучения ее пространственных форм [8]. В работе
[4] была предложена математическая
модель пространственной структуры молекулы РНК, в которой молекулярная цепь
рассматривается как тонкий упругий стержень, имеющий в свободном состоянии
форму винтовой линии. (Свободное состояние означает отсутствие действия на
стержень каких-либо внешних сил и моментов.)
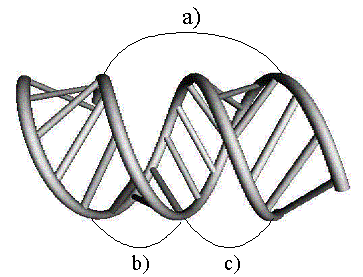
Рис.
9.2. Двойная спираль в A-форме;
a)
шаг витка b) малая бороздка, c) большая бороздка.
С шагом,
равным длине одного нуклеотида, в стержень вделаны жесткие перемычки, равные по
длине половине Уотсон-Криковской связи. В простейшей модели параметры винтовой
линии и ориентация перемычек выбираются так, что два свободных стержня
одинаковой длины, будучи правильно расположены, образуют двойную спираль в
A-форме.
Пространственная структура молекулы собирается из
стеблей и петель в соответствии с заданной вторичной структурой. Каждая петля состоит из семейства тонких
упругих стержней со взаимно согласованными краевыми условиями равновесия. В
соответствии с краевыми условиями концы стержней ориентируются так же, как
концы нитей в стебле А-формы РНК.
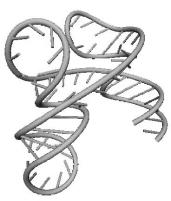
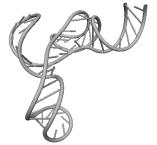
Рис. 9.3. Пример построенной третичной структуры транспортной
РНК
Аспарагиновой кислоты Asterina Pectini.
Пространственная структура
молекулы строится в два этапа. Сначала
определяется ее вторичная структура, а затем третичная.
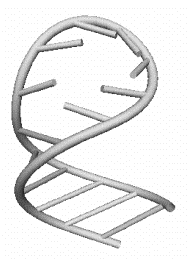
a)
b)
Рис 9.4. Элементы
третичной структуры РНК: a) стебель; b) шпилечная петля
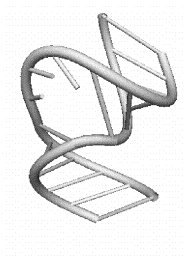
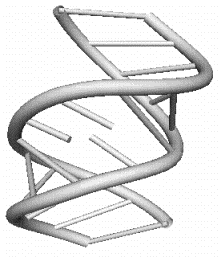
c)
d)
Рис 9.4. Элементы третичной структуры РНК: c) боковая петля; d) внутренняя петля
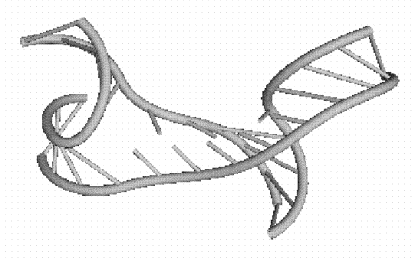
e)
Рис 9.4. Элементы третичной
структуры РНК: e) многозвенная петля.
При
построении третичной структуры молекулярная цепь рассматривается как тонкий
упругий стержень с поперечными стяжками, соответствующими вторичной структуре.
При соответствующем выборе параметров стержня его пространственная форма приближенно описывает пространственную
структуру молекулы РНК. В нашей работе используется одна из наиболее простых
моделей описания пространственных структур молекул РНК - непрерывная модель. В
данной модели молекула РНК описывается как упругий стержень с абсолютно
твердыми поперечными связями. Пространственная структура молекулы собирается из
базовых элементов, каждый из которых является замкнутым контуром, состоящим из
семейства упругих стержней, последовательно соединенных между собой жесткими
перемычками. Определение статически устойчивой пространственной формы базовых
элементов сводится к решению системы краевых задач, каждая из которых состоит в
определении конфигурации тонкого упругого стержня, удовлетворяющей
геометрическим условиям на его концах.
Пространственная форма
стержня определяется на основе уравнений равновесия. Они включают в себя 6
параметров: A, B, C – два главных изгибных
и один крутильный коэффициенты упругости стержня; ![]() ,
, ![]() ,
, ![]() – геометрические
параметры стержня в свободном состоянии (кривизна-кручение в проекции на
главные оси тензора упругости).
– геометрические
параметры стержня в свободном состоянии (кривизна-кручение в проекции на
главные оси тензора упругости).
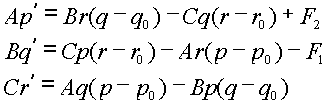
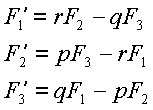
В положении равновесия достигает экстремума
упругая энергия
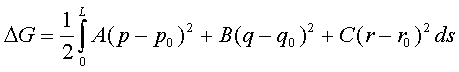
10.
Определение пространственной формы шпилечной петли
Тонкий
упругий стержень, моделирующий шпилечную петлю, является гладким продолжением
одной из нитей двуспирального участка и гладко же переходит другим концом в
другую его нить. Поэтому начальное положение и ориентация стержня совпадают с
конечным положением и ориентацией первой нити двуспирального участка, а
конечное положение и ориентация с начальным положением и ориентацией второй
нити. Параметры двуспирального участка в наших экспериментах совпадали с параметрами
A-формы двойной спирали.
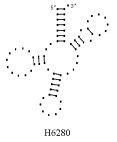
На Рис. 10.1. показана
постановка краевой задачи для шпилечной петли. Поясним обозначения. ![]() есть радиус-вектор осевой линии стержня, параметризованный
ее длиной
есть радиус-вектор осевой линии стержня, параметризованный
ее длиной ![]() ,
, ![]() , i=1,2,3
– направляющие главных осей тензора упругости.
, i=1,2,3
– направляющие главных осей тензора упругости.
![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]()
Угол ![]() – это угол
поворота главных осей тензора упругости относительно осей естественного
трехгранника Френе.
– это угол
поворота главных осей тензора упругости относительно осей естественного
трехгранника Френе.
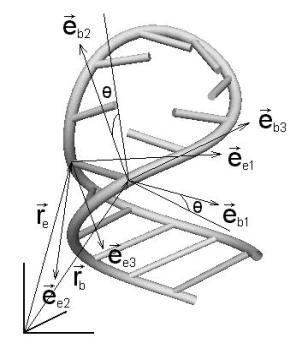
Рис.10.1. Задание краевых условий (для
шпилечной петли). Пространственная структура собирается из стеблей и петель в
соответствии с заданной вторичной структурой.
Каждая петля состоит из семейства тонких упругих стержней со взаимно
согласованными краевыми условиями равновесия. В соответствии с краевыми
условиями концы стержней ориентируются так же, как концы нитей в стебле А-формы
РНК.
11.
Определение формы многозвенных петель
Многозвенная
петля описывается как равновесная форма нескольких участков тонкого упругого
стержня, соединенных твердыми перемычками, моделирующими Уотсон-Криковские
связи. В предыдущем разделе был описан процесс вычисления начальных условий
уравнений равновесия каждого следующего участка по финальным значениям соответствующих
величин для предыдущего участка упругого стержня. Это позволяет поставить
краевую задачу для многозвенной петли подобно тому, как она ставилась для
однозвенной шпилечной петли.
Пусть
в многозвенной петле всего n однонитевых участков, длины которых
обозначаются ![]() . При построении
пространственной формы приходится отыскивать форму многозвенного стержня, для
которого заданы положение и ориентация на левом конце первого участка и на
правом конце последнего участка, а силы и моменты на левом конце неизвестны. Их
приходится определять в ходе решения краевой задачи. Более точно – краевые
условия означают задание следующих величин:
. При построении
пространственной формы приходится отыскивать форму многозвенного стержня, для
которого заданы положение и ориентация на левом конце первого участка и на
правом конце последнего участка, а силы и моменты на левом конце неизвестны. Их
приходится определять в ходе решения краевой задачи. Более точно – краевые
условия означают задание следующих величин:
![]() ,
, ![]() ,
, ![]() (11.1)
(11.1)
![]() ,
, ![]() ,
, ![]()
(верхний индекс здесь – это номер
однонитевого участка). А решение краевой задачи означает правильный выбор
величин
![]() ,
, ![]()
Пространственная форма
молекулы, рассчитанная на основе наших моделей, воспроизводит топологию
пространственной структуры, полученной
методами рентгеноструктурного
анализа. Предлагаемый подход позволяет вычислять пространственные
структуры молекул РНК, для которых не проводился рентгеноструктурный анализ.
12.
Оценка числа стеблей.
Попробуем оценить, каково количество стеблей,
которые могут возникать в молекулярной цепи, первичная структура которой (т.е.
нуклеотидный состав) случайна. Обозначим ![]() – среднее число
стеблей длины
– среднее число
стеблей длины ![]() при случайном выборе
молекулярной цепи, состоящей из
при случайном выборе
молекулярной цепи, состоящей из![]() нуклеотидов.
нуклеотидов.
Оценим сначала количество стеблей длины 1,
т.е стеблей, содержащих только одну Уотсон-Криковскую связь. Если не учитывать
правило комплементарности, то связь, входящая в стебель, может соединять любую
пару нуклеотидов в цепи, а число пар – это ![]() . Введем теперь в рассмотрение
правило комплементарности нуклеотидов. Пусть на левом конце связи, входящей в
стебель, оказался какой-то нуклеотид
. Введем теперь в рассмотрение
правило комплементарности нуклеотидов. Пусть на левом конце связи, входящей в
стебель, оказался какой-то нуклеотид ![]() . Нуклеотид на правом конце может оказаться любым. В
сооответствии с правилом комплементарности, вероятность того, что этот
(случайный) нуклеотид будет комплементарен нуклеотиду
. Нуклеотид на правом конце может оказаться любым. В
сооответствии с правилом комплементарности, вероятность того, что этот
(случайный) нуклеотид будет комплементарен нуклеотиду ![]() , равна
, равна ![]() . Поэтому среднее число стеблей длины 1 в случайной молекулярной
цепи длины
. Поэтому среднее число стеблей длины 1 в случайной молекулярной
цепи длины ![]() оценивается как
оценивается как
![]()
Рассмотрим теперь общий
случай. Расположим нуклеотиды молекулярной цепи последовательно на окружности.
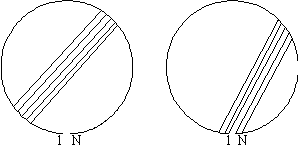
a)
b)
Рис 12.1. Стебель
длины ![]() – это семейство
– это семейство ![]() параллельных, идущих подряд хорд (a). Если в семейство
входят две хорды с концами в точках
параллельных, идущих подряд хорд (a). Если в семейство
входят две хорды с концами в точках ![]() и
и ![]() ,
то это два стебля меньшей длины.
,
то это два стебля меньшей длины.
При таком описании, стебель – это семейство ![]() параллельных хорд,
концы которых сдвинуты на один нуклеотид по отношению к соседним хордам (см.
Рис 12.1. a). Оценим число таких
семейств. Самый левый нуклеотид стебля может занимать
параллельных хорд,
концы которых сдвинуты на один нуклеотид по отношению к соседним хордам (см.
Рис 12.1. a). Оценим число таких
семейств. Самый левый нуклеотид стебля может занимать ![]() положений на
окружности. Допустим, что мы зафиксировали левый конец стебля в нуклеотиде
положений на
окружности. Допустим, что мы зафиксировали левый конец стебля в нуклеотиде ![]() , тогда нуклеотиды m, m+1,…, m+n–1 будут заняты и на окружности останется
, тогда нуклеотиды m, m+1,…, m+n–1 будут заняты и на окружности останется ![]() свободных нуклеотидов
(сложение здесь и ниже производится по модулю N). Если правый конец стебля помещен в нуклеотид j, то нуклеотиды j,
свободных нуклеотидов
(сложение здесь и ниже производится по модулю N). Если правый конец стебля помещен в нуклеотид j, то нуклеотиды j, ![]() ,…,
,…, ![]() будут заняты. Поскольку левые и
правые концы связей стебля не должны попадать в одни и те же позиции, то для
правого конца стебля остается
будут заняты. Поскольку левые и
правые концы связей стебля не должны попадать в одни и те же позиции, то для
правого конца стебля остается ![]() возможных положений.
Это
возможных положений.
Это ![]() ,
, ![]() ,…,
,…, ![]() . Таким образом, всего имеется
. Таким образом, всего имеется ![]() семейств параллельных
хорд, но эту величину надо еще уменьшить вдвое, поскольку каждое семейство
участвовало в нашем подсчете дважды – мы можем назвать левой стороной семейства
хорд любую из его двух сторон. Итак, всего мы можем получить
семейств параллельных
хорд, но эту величину надо еще уменьшить вдвое, поскольку каждое семейство
участвовало в нашем подсчете дважды – мы можем назвать левой стороной семейства
хорд любую из его двух сторон. Итак, всего мы можем получить
![]()
семейств n параллельных хорд,
концы которых сдвинуты на один нуклеотид по отношению к соседним хордам.
Найденная величина r включает в себя не только семейства хорд,
описывающих стебли длины n, но и неправильные семейства, т.е семейства, описывающие
два стебля меньшей длины (см. Рис 12.1. b). Это семейства,
содержащие две хорды с концами в точках ![]() и N. Левый конец таких семейств может занимать
и N. Левый конец таких семейств может занимать ![]() положение, располагаясь
в позициях N,
положение, располагаясь
в позициях N, ![]() ,…,
,…, ![]() . Правый конец семейства (как и в общем случае) может
занимать
. Правый конец семейства (как и в общем случае) может
занимать ![]() положений. И всего (с
учетом двукратного участия семейств хорд в нашем рассмотрении) мы получим
положений. И всего (с
учетом двукратного участия семейств хорд в нашем рассмотрении) мы получим
![]()
неправильных семейств хорд.
Вычитая неправильные
семейства из общего количества, получим,
что общее количество правильных семейств хорд равно ![]() . Учитывая, что вероятность комплементарности оснований во
всех n связях стебля равна
. Учитывая, что вероятность комплементарности оснований во
всех n связях стебля равна ![]() , получим следующую оценку среднего числа стеблей длины
n в молекулярной цепи из N случайно выбранных нуклеотидов:
, получим следующую оценку среднего числа стеблей длины
n в молекулярной цепи из N случайно выбранных нуклеотидов:
![]()
Можно оценить и общее
количество стеблей любой возможной длины. Обозначим эту величину ![]() . Она равна сумме
. Она равна сумме ![]() по всем возможным n. С одной стороны
по всем возможным n. С одной стороны ![]() не меньше, чем
не меньше, чем ![]() . С другой стороны
. С другой стороны ![]() , а
, а ![]() . Поэтому суммирование величин
. Поэтому суммирование величин ![]() по всем возможным n даст величину меньшую, чем
по всем возможным n даст величину меньшую, чем ![]() .
.
Таким образом, скорость роста общего
количества стеблей ![]() при увеличении длины
молекулярной цепи
при увеличении длины
молекулярной цепи ![]() имеет порядок не
меньше
имеет порядок не
меньше ![]() и не больше
и не больше ![]() .
.
13.
Оценка числа структур.
Попробуем оценить, каково количество
вторичных структур, которые могут возникать в молекулярной цепи, первичная
структура которой (т.е. нуклеотидный состав) случайна.
Будем называть длиной структуры количество Уотсон-Криковских связей в ней. Будем
рассматривать только структуры, удовлетворяющие стерическому условию. Обозначим
![]() – среднее число
структур длины
– среднее число
структур длины ![]() при случайном выборе
молекулярной цепи, состоящей из
при случайном выборе
молекулярной цепи, состоящей из![]() нуклеотидов.
нуклеотидов.
Количество структур длины 1 равно числу
стеблей, содержащих только одну Уотсон-Криковскую связь:
![]()
Рассмотрим теперь структуру длины n. В ней какие-то ![]() нуклеотидов попарно
связаны Уотсон-Криковскими связями. Всего возможно
нуклеотидов попарно
связаны Уотсон-Криковскими связями. Всего возможно ![]() различных выборов
различных выборов ![]() нуклеотидов. После того, как нуклеотиды выбраны, вторичная
структура определяется системой связей между ними. Перенумеруем выбранные
нуклеотиды последовательно от 1 до
нуклеотидов. После того, как нуклеотиды выбраны, вторичная
структура определяется системой связей между ними. Перенумеруем выбранные
нуклеотиды последовательно от 1 до ![]() и расположим их в
вершинах правильного
и расположим их в
вершинах правильного ![]() -угольника. Соединим связанные нуклеотиды диагоналями. Каждой вторичной структуре взаимно
однозначно соответствует набор из
-угольника. Соединим связанные нуклеотиды диагоналями. Каждой вторичной структуре взаимно
однозначно соответствует набор из ![]() непересекающихся
диагоналей.
непересекающихся
диагоналей.
Обозначим ![]() число различных
расположений
число различных
расположений ![]() непересекающихся диагоналей в правильном
непересекающихся диагоналей в правильном ![]() -угольнике. Тогда (с учетом того, что вероятность
комплементарности выбранных нуклеотидов равна
-угольнике. Тогда (с учетом того, что вероятность
комплементарности выбранных нуклеотидов равна ![]() ) имеем
) имеем

Поскольку ![]() , то
, то
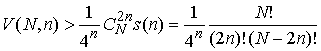 ,
,
причём степень полинома от ![]() , стоящего а правой части, равна
, стоящего а правой части, равна ![]() . Отсюда можно сделать два вывода:
. Отсюда можно сделать два вывода:
·
С ростом молекулярной цепи количество возможных вторичных структур,
состоящих из ![]() Уотсон-Криковских
связей, растет как
Уотсон-Криковских
связей, растет как ![]() .
.
·
С ростом молекулярной цепи количество всех возможных вторичных структур
растет быстрее чем любой полином (иначе говоря скорость роста количества
вторичных структур больше полиномиальной).
Несложно убедиться,
что ![]() ,
, ![]() ,
, ![]() . Однако общая формула для
. Однако общая формула для ![]() неизвестна.
неизвестна.
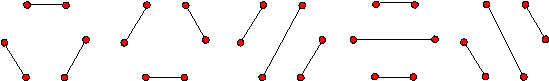
Рис. 13.1. Пять вариантов
вторичной структуры с тремя связями (![]() ).
).
Рассмотрев структуры, у которых связаны
нуклеотиды 1 и 2, а также структуры, у которых связаны нуклеотиды 2 и 3,
несложно убедиться, что ![]() . Отсюда получаем, что
. Отсюда получаем, что ![]() . Следовательно, для среднего числа структур длины
. Следовательно, для среднего числа структур длины ![]() при случайном выборе
молекулярной цепи, состоящей из
при случайном выборе
молекулярной цепи, состоящей из ![]() нуклеотидов, верна оценка
нуклеотидов, верна оценка
![]()
14.
Оценка
вычислительной сложности задачи.
В ходе транскрипции молекулярная цепь РНК
последовательно удлиняется с некоторой скоростью. При этом на участке уже
выросшей цепи могут образовываться водородные связи между комплементарными
нуклеотидами. Совокупность этих связей называется вторичной структурой
молекулы. Она, в свою очередь, определяет энергетически более слабые
пространственные изгибы молекулярной цепи. В ходе роста молекулярной цепи могут возникать новые вторичные связи
или уже имеющиеся вторичные связи могут разрываться и замещаться энергетически
более выгодными комбинациями других вторичных
связей (перестройка структуры). Поскольку
число возможных вторичных структур конечно (имеет порядок куба от числа
нуклеотидов в молекулярной цепи), то процесс формирования или перестройки
вторичной структуры молекулы дискретен и эквивалентен переходу от одной вершины
графа возможных вторичных структур к другой.
Множество допустимых переходов определяется моделью процесса. В нашем
случае допускаются только локальные переходы, в ходе которых в структуре могут
возникать новые элементарные подструктуры, т.н. стебли. Любая вторичная
структура может быть описана семейством неперекрывающихся стеблей. Число
возможных стеблей имеет порядок 0.1 от квадрата числа нуклеотидов в молекулярной цепи. Такой же порядок имеет и число
допустимых переходов между соседними вершинами графа допустимых вторичных
структур. Отметим, что длина цепи РНК может варьироваться от десятков до
десятков тысяч нуклеотидов. Основное время вычислительного процесса при
компьютерном моделировании расходуется на выбор ребра перехода от текущей
вершины графа вторичных структур к следующей, обеспечивающего минимизацию свободной
энергии структуры на множестве допустимых переходов. Этот же этап
вычислительного процесса наиболее просто перелагается на параллельные
вычислительные устройства. Оценим эффективность такого распареллеливания. Общее
время, затрачиваемое одним процессором на поиск стебля, добавление которого к
имеющейся структуре понижает свободную энергию молекулы наибольшим образом,
оценивается как
![]() ,
,
где ![]() – время вычисления свободной энергии новой
структуры,
– время вычисления свободной энергии новой
структуры, ![]() - число допустимых переходов (стеблей). Величина
- число допустимых переходов (стеблей). Величина
![]() линейно растет с длиной молекулярной цепи, а
линейно растет с длиной молекулярной цепи, а ![]() , как уже
говорилось - квадратично, поэтому
, как уже
говорилось - квадратично, поэтому ![]() , где
, где ![]() - число нуклеотидов в молекулярной цепи, а
- число нуклеотидов в молекулярной цепи, а ![]() - коэффициент пропорциональности. Если
использовать вычислительную систему с
- коэффициент пропорциональности. Если
использовать вычислительную систему с ![]() процессорами, то время вычислений сократится
в
процессорами, то время вычислений сократится
в ![]() раз, но возникнут потери, связанные с
пересылкой данных между процессорами. Пересылаемая информация - это текущая вторичная структура (она имеет
порядок
раз, но возникнут потери, связанные с
пересылкой данных между процессорами. Пересылаемая информация - это текущая вторичная структура (она имеет
порядок ![]() ) и выбранный
стебель из
) и выбранный
стебель из ![]() -ой части
допустимых стеблей (объем этой информации примерно постоянен).
-ой части
допустимых стеблей (объем этой информации примерно постоянен).
Таким образом, общее время вычислений в
параллельном случае можно оценить как
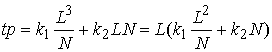
Нетрудно видеть, что эта величина имеет минимум по N, равный ![]() , и достигающийся
при
, и достигающийся
при 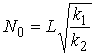 . Это означает, что с ростом числа процессоров на параллельной
системе можно понизить время вычислений с
. Это означает, что с ростом числа процессоров на параллельной
системе можно понизить время вычислений с ![]() до
до ![]() , при этом оптимальное
число процессоров имеет порядок
, при этом оптимальное
число процессоров имеет порядок ![]() .
.
Рассмотрим теперь реальную ситуацию, когда у нас
есть ограниченное число процессоров (N), но мы работаем со все более длинными молекулами. В этом случае
эффективность распаралеливания (оцениваемая как ![]() ) меняется
следующим образом:
) меняется
следующим образом:
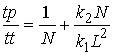
Ясно, что с ростом длины молекулярной цепи (![]() ) эффективность
стремится к
) эффективность
стремится к ![]() , т.е. к
максимально теоретически возможной. Следовательно, в случае нашей задачи на
паралельных системах выгодно моделировать структурообразование длинных молекул,
что коррелирует с тем фактом, что
структура именно длинных молекул наболее тяжело раскрывается другими
методами.
, т.е. к
максимально теоретически возможной. Следовательно, в случае нашей задачи на
паралельных системах выгодно моделировать структурообразование длинных молекул,
что коррелирует с тем фактом, что
структура именно длинных молекул наболее тяжело раскрывается другими
методами.
15.
Вычислительные
эксперименты на параллельных системах.
Выше мы
показали, что вычислительная сложность моделирования процесса
образования вторичных структур РНК при заданных параметрах процесса имеет кубический рост в зависимости от длины молекулы. При этом квадрат времени
раходуется на вычисления свбодной энергии переходов от текущей структуры к потенциально возможным
перестроенным структурам. В то же время
особый интерес представляет изучение структурообразования длинных молекул. Заметим, что расчеты энергии структурных переходов
могут вестись независмо друг от друга. Это определяет перспективность использования
многопроцессорных систем для данного круга задач.
В 1995 году нами был разработан программный
комплекс GEN, который позволяет проводить исследования процессов
образования вторичной структуры РНК на
многопроцессорном комплексе МВС-100. Он позволяет исследовать характеристики этого процесса для заданного
семейства молекул. Распараллеливание
процесса молелирования происходит на уровне расчета энергий возможных
межструктурных переходов. Количество таких
переходов для молекул длиной до 100 нуклеотидов имеет порядок нескольких
тысяч. Для молекул до 500 нуклеотидов – несколько десятков тысяч. А для длины
1000 - 2000 – порядка сотен тысяч - миллиона. Это позволяет эффективно
использовать возможности
многопроцессорного комплекса.
a)
b)
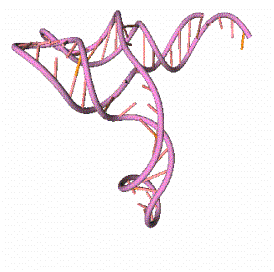
c)
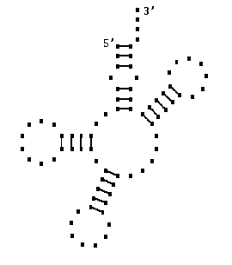
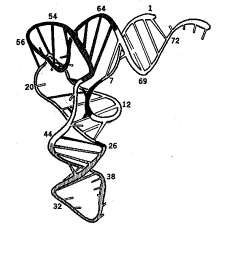
Рис. 15.1. Пример пространственной структуры молекулы РНК,
полученной методами математического моделирования. Пространственная структура
молекулы тРНК Phenilalanine Yeast. a) Вторичная
структура, b) Рентгенострукутрный анализ [5]. c) Компьютерные
вычисления.
В 1995-97 годах на программном комплексе GEN были исследованы процессы структурообразования для интересных
классов молекул РНК - транспортных (тРНК), рибосомальных (5S РНК), а также недавно открытых
молекул-ферментов - рибонуклеаза P-РНК [6]. Длина этих молекул составляет
300 – 400 нуклеотидов. В семейство входит семь экземпляров молекул. Общее
время расчета составило 317 часов, при этом число процессоров, участвующих в
расчете, менялось в зависмости от аппаратных ресурсов, которые предоставлялись
программе - от одного до 32. Заметим,
что примерное время такого расчета с использованием только одного
процессора составило бы несколько тысч часов.
Эксперименты с комплексом GEN показали высокую эффективость использования многопроцессорных методов
вычислений в задачах исследования структурообразования биологических
макроморлекул. Оказалось, что для
молекул длиной до 500 нуклеотидов эффективность распараллеливания составляет 60
- 70%. В настоящее время начаты
работы по развитию следующего варианта
комплекса GEN с целью
увеличить возможную длину иссследумых молекул до 3000 нуклеотидов. При
исследовании молекул такой длины эффективность параллельной работы процессоров должна составить 90% и выше.
В 1997-2000 годах комплекс GEN был развит, и на его основе был создан
комплекс GEN-2, который
позволяет исследовать характеристики структурообразования молекул РНК длиной до
3000 нуклеотидов. В настоящее время комплекс GEN-2 прошел проверку и на нем проводятся исследования процесса структурообразования
молекул из класса рибосомальных субъединиц 16S PHK. Средняя длина таких
молекул составляет 2000 нуклеотидов.
16.
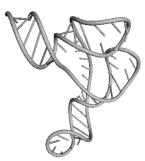
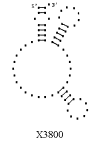
Примеры структур тРНК.
В данном разделе приводятся примеры третичных структур транспортных РНК,
полученных методами, описанными в данной работе.
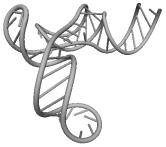
UGC THERMOPROT. TENAX GCA RAT
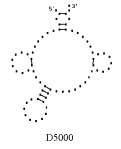
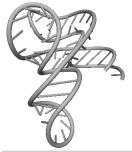
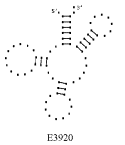
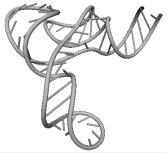
GUC ASTERINA PECTINI. UUC NICOTIANA TABACUM
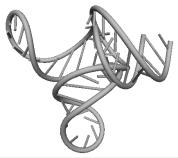
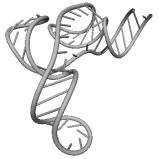
GAA ASCARIS SUUM UCC CHLAMYDOMO. REINH.
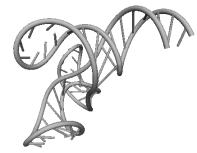

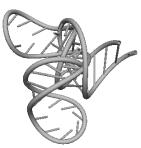
GUG SACCHAROMYCES CER. GAU ASCARIS SUUM
CUU TRYPANOSOMA BRUCEI UAG EUGLENA GRACILIS
CAU HORDEUM VULGARE GUU TRITICUM AESTIVUM
GGG MARCHANTIA POLYM. UUG HUMAN
ACG CAENORHABDI. ELEG. UGA NICOTIANA TABACUM

UGU BACILLUS SUBTILIS UAC ASCARIS SUUM

CCA SACCHAROMYCES CER. CAU TETRAHYMENA PYRIF.
Вторичные структуры молекул были взяты из [7].
Молекулы выбирались случайным образом – по одной для каждой аминокислоты.
Литература
1.
Энеев Т.М.,
Козлов Н.Н., Кугушев Е.И. Процессы структуризации биомолекул. Результаты
математического моделирования. Препринт
ИПМ им. М.В. Келдыша РАН, N
69, 1995, с. 22.
2.
Козлов Н.Н.,
Кугушев Е.И., Энеев Т.М. Структурообразующие
характеристики транскрипционного процесса. Математическое моделирование т.10, N 6, с.3-19, 1998.
3.
Козлов Н.Н.,
Кугушев Е.И., Энеев Т.М. Параллельные вычисления при решении некоторых задач
астрофизики и молекулярной биологии. Математическое моделирование т.12, N 7, с.65-70, 2000.
4.
Кугушев
Е.И., Старостин Е.Л., Пирогова Е.Е.
Математическая модель образования трехмерной структуры РНК. Препринт ИПМ им. М.В.
Келдыша РАН, N 77, 1997, с.
24
5. Kim S.H., Suddath F.L., Qugley G.J., McPherson A., Sussman J.L., Wang A.H.J., Seeman N.C., Rich A. (1974) Three-Dimensional Tertiary Structure of Yeast Phenylalanine Transfer RNA. Science, 185, N 4149, 435-440.
6. S.-Y. Lee and M. Zuker, Jorn. of Biomol. Struct. and Dynamics. v.8, N5, 1991 pp. 1027- 1044.
7.
Sprinzl M., Dank N., Nock S., Schon A. Compilation of tRNA
sequnces and sequences of tRNA genes. // Nucleic Acids Res., 1991, v. 19, suppl., p. 2127-2171.
8.
Benham C.J. Geometry and mechanics of DNA superhelicity. Biopolymers,
1983, v. 22, N 11, pp 2477-2495.